Research


UNBC NRESi Biodiversity monitoring & Assessment program (BMAP)
I am a part of a team of UNBC researchers that aim to provide best practice recommendations for operational practices associated with the construction of a natural gas pipeline. My focus will be on identifying restoration treatments that favor the movement of small mammals along an across the corridor created by the pipeline right-of-way. We are currently developing an optimized noninvasive sampling approach that will target a number of small/meso forest dwelling mammals.
Collaborators:
Jeanne Robert, UNBC BMAP research manager.
Animal movement - Mike Gillingham, Chris Johnson, Ken Otter, Roy Rea, (Co-PIs UNBC), David Graham and Alexander LePage (UNBC research assistants)
Invasive plants - Lito Arocena (PI, UNBC), Mike Rutherford, Keith Egger, Chris Opio, and Hugues Massicotte (Co-PIs UNBC)
Population genetics of invasive plants
Invasive alien species have been introduced to new area for as long as humans have traveled and have become one of the most important cause for the loss of biodiversity worldwide. In order to better understand why certain species spread and become invasive, I am interested in studying population genetics and invasion history of invasive organisms. During my MSc I had the opportunity to work on Invasive Olives (Olea europeae) from Australia and Hawaii (see Besnard et al., 2007a, 2007b). I then focused on Invasive Giant Hogweed (Heracleum mantegazzianum) in the western Swiss Alps. This plant was introduced to temperate areas of the world through the horticultural industry due to its beautiful and imposing inflorescences. It has now established invasive populations in over 20 European countries as well as in the USA and Canada. Besides being a treat to native flora, Giant Hogweed is a toxic plant that is becoming a health hazard for people living in invaded areas. Such studies using the tools of population genetics of invasive plants will help identify invasion pathways as well as the main dispersal agents (roads, rivers, etc) and will ultimately be useful in establishing an eradication plans for such undesirable species. (See Henry et al., 2008 for primer design and Henry et al., 2009 for population genetic study). An ongoing collaboration seeks to begin to shed light on the genetic basis of invasiveness in the Giant Hogweed by using genome scans and samples from throughout native and invaded range.
Collaborators:
Guillaume Besnard (CNRS Toulouse, FR), Gwenaelle Le Lay (Unil, CH), Antoine Guisan (Unil, CH), Jerome Goudet (Unil, CH), Sarka Jahodova (Charles University, Prague, CZ) and Jim Provan (Queens University, Belfast, UK)
In situ and Ex situ conservation genetics of Amur tigers (Panthera tigris altaica)
The Amur or Siberian Tiger is a critically endangered felid. Due to human pressures, its population plummetted to 20-30 individuals in the 1940s. Conservation efforts in the wild have enabled the population to rebound to an estimated 500 individuals. In the meantime, a captive population was set up mainly in European and North American Zoo (with a total of 421 individuals). This project aims to compare the amount of genetic variability remaining in the wild compared to the captive population. Moreover, we are interested in the demographic history of the wild population and will investigate how a development corridor between two cities in the Russian Far East has affected the population genetic structure of the remaining wild Amur tiger population. In our recently published work (Henry et al., 2009) we found a low effective population size in the wild Amur tiger of 35 individuals. We also discovered that this population was split into two by a development corridor between the cities of Vladivostok and Ussurisk. This has important implication for the future conservation measures that should focus on creating forest corridors to enable the tigers to move from one population to the next. Moreover we found that the captive breeding program has successfully maintained genetic diversity comparable to that found in the wild.
Collaborators:
Taro Sugimoto (Hokaido University, JP), Dale Miquelle (WCS), Dale McCullough (UC Berkley), Gisella Caccone (Yale University, USA) and Michael Russello (PI; UBC O)
Genetic basis of adaptation in a conservation context
A major gap in conservation genetics has been to identify genomic regions that display signals of positive selection and can thus be use to prioritize population for conservation action and/or assisted migration. I have thus been investigating the adaptive genetic variation in the American Pika (Ochotona princeps) using emerging tools of conservation genomics. I have developed a noninvasive sampling technique well suited to the species in order to obtain large quantities of their hair that would provide the DNA for my study (Henry, Henry & Russello, 2011 and Henry & Russello 2011). American Pikas have recently been identified as an early warning for climate change in mountainous regions of North America. We have sampled Pika populations along three altitudinal gradients in the Coast Mountains of BC (Henry, SIm & Russello, 2012), and have investigated how these populations differ at the genomic level. This entailed using AFLP genomic scans to identify outlier loci (areas of the genome exhibiting signatures of selection) in pika populations along the altitudinal gradients. This study will represent one of the first applications of population genomics in a species of conservation concern (Henry & Russello, 2013). In addition to the work undertaken as part of my PhD thesis, we are currently typing the above populations at genome wide SNPs and will extend the sampling efforts in order to test for genomic signals of selection along latitudinal gradients ranging from Oregon to central Alberta and British Columbia and habitat characteristics of pika populations along an elevation gradient.
Collaborators:
Michael Russello (PhD Advisor, UBC O), Kelsey Robson (MSc student UBC O), Karen Hodges (Committee member, UBC O), Michael Pidwirny (Committee member, UBC O), Mark Rheault (Committee member, UBC O) and Karl Larsen (Committee member, TRU), David Hik ( External examiner, U of A), Jesse Whittington, Derek Peterson and Julie TImmins (Parks Canada).

Giant Hogweed (Heracleum mantegazzianum) in the western Swiss Alps. Photo courtesy of Florian Dessimoz
Links: Giant Hogweed working group, University of Lausanne, Switzerland (In French)

Amur tigers (Panthera tigris altaica) at the Buffalo Zoo. Photo courtesy of Dave Pape.
Links: Media coverage by the BBC

An American pika (Ochotona princeps) emerges from the boulders at Rock Glacier, Kananaskis country, Alberta. Photo courtesy of Alison Henry.
Recent contributions
-
-Henry P, Hilyard A, Johnson S, Orser C. (2018) Predicting chemovar cluster and variety verification in vegetative cannabis accessions using targeted single nucleotide polymorphisms. PeerJ Preprints 6:e27442v1 https://doi.org/10.7287/peerj.preprints.27442v1
-
-Henry P. (2018) Two Single Nucleotide Polymorphisms Tease Apart Hemp, CBDA and THCA Expressing Cannabis. The Journal of Brief Ideas http://beta.briefideas.org/ideas/199d82d72bf64234b8ace849cc20896c
- Henry P. (2018) The genetic basis of the human-cannabis relationship. BioRxiv 2887938; doi: https://doi.org/10.1101/287938
-
-Henry P. (2017) Cannabis chemovar classification: terpenes hyper-classes and targeted genetic markers for accurate discrimination of flavours and effects. PeerJ Preprints5:e3307v1 https://doi.org/10.7287/peerj.preprints.3307v1
-
-Henry, P. (2017): Discordant signals of genetic differentiation in the Cannabis genus based on SNP and SSR data. figshare. https://doi.org/10.6084/m9.figshare.4967765.v1
-
-Henry P. and Wille L. Referee Report For: Metagenomic analysis of medicinal Cannabis samples; pathogenic bacteria, toxigenic fungi, and beneficial microbes grow in culture-based yeast and mold tests. F1000Research 2016, 5:2471 (http://f1000research.com/articles/5-2471/v1#referee-response-16859)
-
-Henry P. The cannabis-human relationship [v1; not peer reviewed]. F1000Research 2016, 5:1986 (slides) (doi: 10.7490/f1000research.1112841.1)
-
-Henry P. (2016) Cannabis and human creativity: hyper-priming, eureka moments and intermittent abstinence. The Journal of Brief Ideas. http://dx.doi.org/10.5281/zenodo.51795
- Henry P. (2016) On Domestication and Feralization: re-rethinking invasive species from a genetic perspective. The Journal of Brief Ideas. http://beta.briefideas.org/ideas/1a766837d477249d9af2f53638baf08a
- Henry P. (2016) Applying Genomic tools to Cannabis Breeding. The Journal of Brief Ideas. http://beta.briefideas.org/ideas/46acc95d5b851cf28facc88ff54b30e7
- Robert, J., Henry, P., Gillingham, M. (2016) Field testing the assessment of baseline small mammal biodiversity using hair-snare techniques in combination with metabarcoding using next-generation sequencing. Biodiversity Monitoring and Assessment Program, Final report to Chevron Inc. Chapter 6.3
- Henry P. (2015) Genome-wide analyses reveal clustering in Cannabis cultivars: the ancient trilogy of a panacea. PeerJ PrePrints 3:e1935https://dx.doi.org/10.7287/peerj.preprints.1553
The Recreational Genomics Consortium
Recreational Genomics is a consortium of cannabis aficionados, cannaseurs, breeders and growers involved in developing genomic resources to advance marker assisted as well as genomic selection to understand and enhance the human-cannabis relationship. Identifying markers related to plant vigor, active compound synthesis, pest resistance and coloration are currently being investigated using open access data generated and hosted by NCBI UofT and the Medicinal Genomics Corporation.
420andMe : crowdsourcing the Cannabis / Human relationship
Cannabis (Cannabis sativa L.) is often cited as the most commonly used recreational drug worldwide. The most recent National Survey on Drug Use and Health confirms this assertion, showing that about 34% to 42% of Americans have used cannabis in their lifetime (one-time users), 10% to 12% have used it in the past year (casual users), 6% to 7% are regular users and about 4% are chronic users. While most participants may experience a feeling of relaxation, exuberance, laughter and/or hunger, others may react differently and exhibit social anxiety or other negative responses. Such outcomes may be mediated by individual differences in gene encoding the endocannabinoid system and other associated regulatory enzymes. In this crowdsourced genome-wide association study, I intend to harness the large amounts of data generated by direct-to-consumer genetic testing through a web-based repository platform to collect genotypic and phenotypic data called OpenSNP.


COMT polymorphism and cognitive effects of cannabis use: In the first study using a purely non-clinical cohort, Tunbridge et al. (2015) report on an additional player in the ECS pathways, the Catechol-O-Methyl-Transferase (COMT) gene polymorphism (Val(158)Met). Their data suggests that COMT genotype moderates the cognitive, but not the psychotic, effects of acutely administered THC, with the Met variant, providing protection to impaired cognitive performance experience by participants with Val variants. Collaborators: OpenSNP, Genesforgood, Dr. Natasha Ryz @ UBC, The Healing Company
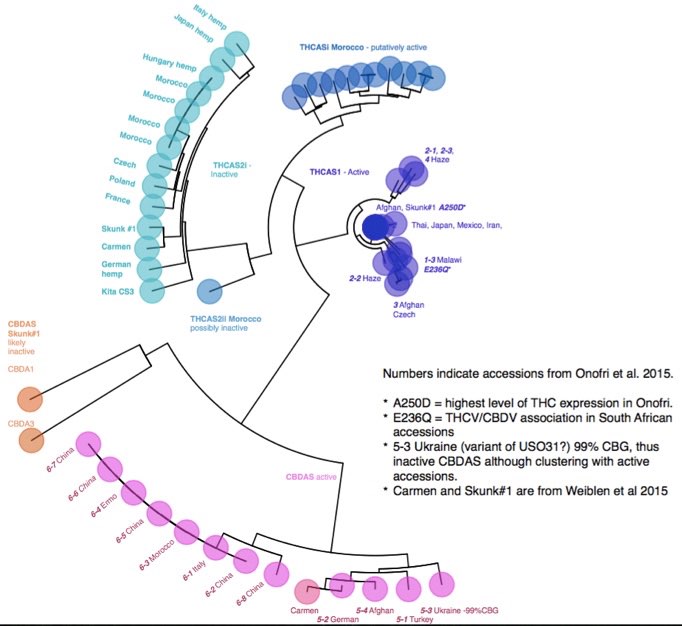
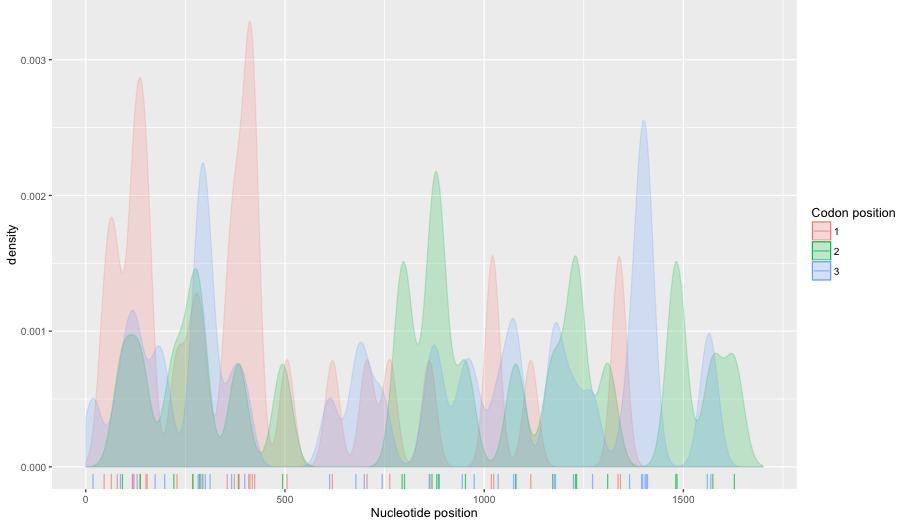
Distribution of SNPs in the THCAS gene based on 58 accessions publicly available on NCBI database
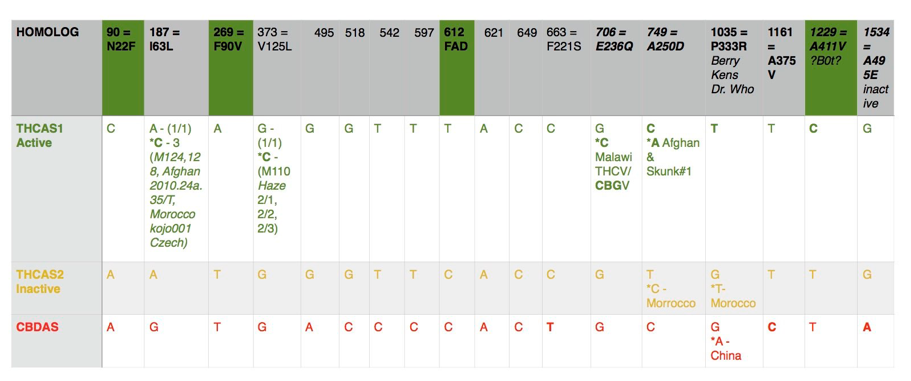
Description of variants in the THCAS homologues and CBDAS gene. Green indicates diagnostic SNPs between active and inactive homologues. Nomenclature follows published work by


